library(data.table)Graficación de GWAS
Durante esta clase veremos como usar data.table para manejar datos de gran tamaño. Por ejemplo los summary statistics de GWAS. Los datos aquí usados se descargaron: Zenodo MXB Y son datos del artículo: Mexican Biobank advances population and medical genomics of diverse ancestries Para hacer este ejercicio sólo utilizaremos el archivo Tryglicerides_INT.imputed.stats.gz que se encuentra dentro de la carpeta full_cohort.
Procesamiento de datos
Cargar librería
Lectura de archivos
Primero leeremos nuestro archivo comprimido, para no ocupar tanto espacio de almacenamiento en nuestras computadoras. También veremos el tiempo que tarda nuestra máquina en leer el archivo.
start.time <- Sys.time()
triglycerides <- fread(file = "/Users/mpalma/Downloads/full_cohort/Glucose_INT.imputed.stats.gz", header = T)
end.time <- Sys.time()Ahora, hagamos una pequeña exploración de nuestro archivo:
head(triglycerides) CHROM GENPOS ID ALLELE0 ALLELE1 A1FREQ INFO N TEST BETA SE CHISQ
<int> <int> <char> <char> <char> <num> <num> <int> <char> <num> <num> <num>
1: 1 10894 chr1:10894:G:A A G 0.998977 0.103625 4418 ADD 0.3059710 0.9390870 0.106157
2: 1 10915 chr1:10915:G:A A G 0.999336 0.143104 4418 ADD 0.3243080 0.9879230 0.107763
3: 1 10930 chr1:10930:G:A A G 0.996291 0.348130 4418 ADD 0.4716960 0.2689780 3.075330
4: 1 10989 chr1:10989:G:A A G 0.996688 0.256007 4418 ADD -0.1462470 0.3317240 0.194366
5: 1 11171 chr1:11171:CCTTG:C C CCTTG 0.947664 0.270913 4418 ADD 0.0323001 0.0822278 0.154301
6: 1 23197 rs1220638906 T TTAAAA 0.992480 0.289927 4418 ADD -0.2011720 0.2076770 0.938331
P LOG10P EXTRA
<num> <num> <lgcl>
1: 0.74456223 0.128099 NA
2: 0.74270612 0.129183 NA
3: 0.07948771 1.099700 NA
4: 0.65930748 0.180912 NA
5: 0.69445802 0.158354 NA
6: 0.33270705 0.477938 NAclass(triglycerides)[1] "data.table" "data.frame"dim(triglycerides)[1] 25454794 15Como podemos ver, tenemos demasiados renglones, más de 25 millones. Las columnas de nuestro archivo:
- CHROM
- GENPOS
- ID
- ALLELE0
- ALLELE1
- A1FREQ
- INFO
- N
- TEST
- BETA
- SE
- CHISQ
- P
- LOG10P
- EXTRA
¿Para nuestro GWAS qué columnas necesitamos?
- CHROM
- GENPOS
- ID
- P
triglycerides <- triglycerides[, .(CHROM, GENPOS, ID, P)]Simplifiquemos los nombres de nuestras columnas
triglycerides <- triglycerides[,.(CHR = CHROM, BP=GENPOS , SNP=ID, P)]Puedes explorar setnames para maneras alternativas de renombrar
Reto: ¿Cómo lo harías?
Consideremos que en el caso de los humanos tenemos 22 cromosomas autosomales y cada variante tiene asociada un cromosoma y una posición en dicho cromosomas.
¿Cómo hacemos que estas posiciones se puedan acomodar en el eje de las x?
Algoritmo
- Agrupar nuestros datos por cromosoma y estimar su longitud
- Hacer la suma acumulada de las posiciones.
Como puedes notar, no tenemos absolutamente todas las posiciones del genoma humano, pero nos es relevante la posición máxima que tenemos en los cromosomas.
Agrupar por cromosoma y estimar la longitud
chr_len <- triglycerides[, .(chr_len = max(BP)), by = CHR]Estimar la suma acumulada de la longitud de cromosomas
chr_len[, tot := cumsum(as.numeric(chr_len)) - chr_len]Añador esta informacio al data set original
cum_triglycerides <- merge(triglycerides, chr_len[, .(CHR, tot)], by = "CHR")Añadir la posicion acumulada a cada variante/SNP (Single Nucleotide Polymorphism)
cum_triglycerides <- cum_triglycerides[order(CHR, BP)]
cum_triglycerides[, BPcum := BP + tot]Todo parece que va muy bien, pero ¿Creen que sea necesario graficar cada variante? ¿Podemos muestrear al azar? Mejor usamos p-value.
Muestrear sin remplazo de manera aleatoria 1 de cada 200 variantes con P-value > 0.01
set.seed(123) # Establecer una semilla para reproducibilidad
sampled_high_p <- cum_triglycerides[P > 0.01][sample(.N, .N / 200)]
low_p_data <- cum_triglycerides[P <= 0.01]
# Combinar ambos conjuntos de datos
sampled_data <- rbind(low_p_data, sampled_high_p)Graficación
Cargar librería
library(ggplot2)
library(scales)Para el eje de las x hay que estimar la posición central para cada cromosoma de acuerdo a las posiciones de cada cromosoma
axisdf <- cum_triglycerides[, .(center = (max(BPcum) + min(BPcum)) / 2), by = CHR]Manhattan plot
Manhattan_plot <- ggplot(sampled_data, aes(x=BPcum, y=-log10(P))) +
# Graficar los puntos y azul y gris intercalado por cromosoma
geom_point( aes(color=as.factor(CHR)), alpha=0.8, size=0.5) +
scale_color_manual(values = rep(c("grey", "skyblue"), 22 )) +
# Personalizar ejes
scale_x_continuous( label = axisdf$CHR, breaks= axisdf$center ) +
scale_y_continuous(expand = c(0, 0), labels=number_format(accuracy = 0.1) , limits=c(0,25)) +
#### Añadir líneas de significancia
geom_hline(yintercept=-log10(1e-5), color = "black", linetype="dotted", linewidth=0.3) +
geom_hline(yintercept=-log10(5e-8), color = "black", linetype="dashed", linewidth=0.3) +
#### Añadir título
ggtitle("Glucose")+
# Detalles de tema:
theme_bw() +
theme(
legend.position="none",
panel.border = element_blank(),
panel.grid.major.x = element_blank(),
panel.grid.minor.x = element_blank(),
#axis.ticks.x = element_blank(),
#axis.text.x = element_blank(),
axis.title.x = element_blank()
)Guardar la imagen
ggsave("/Users/mpalma/Downloads/Manhattan_viernesbioinfo.png", plot = Manhattan_plot)Plot
print(Manhattan_plot)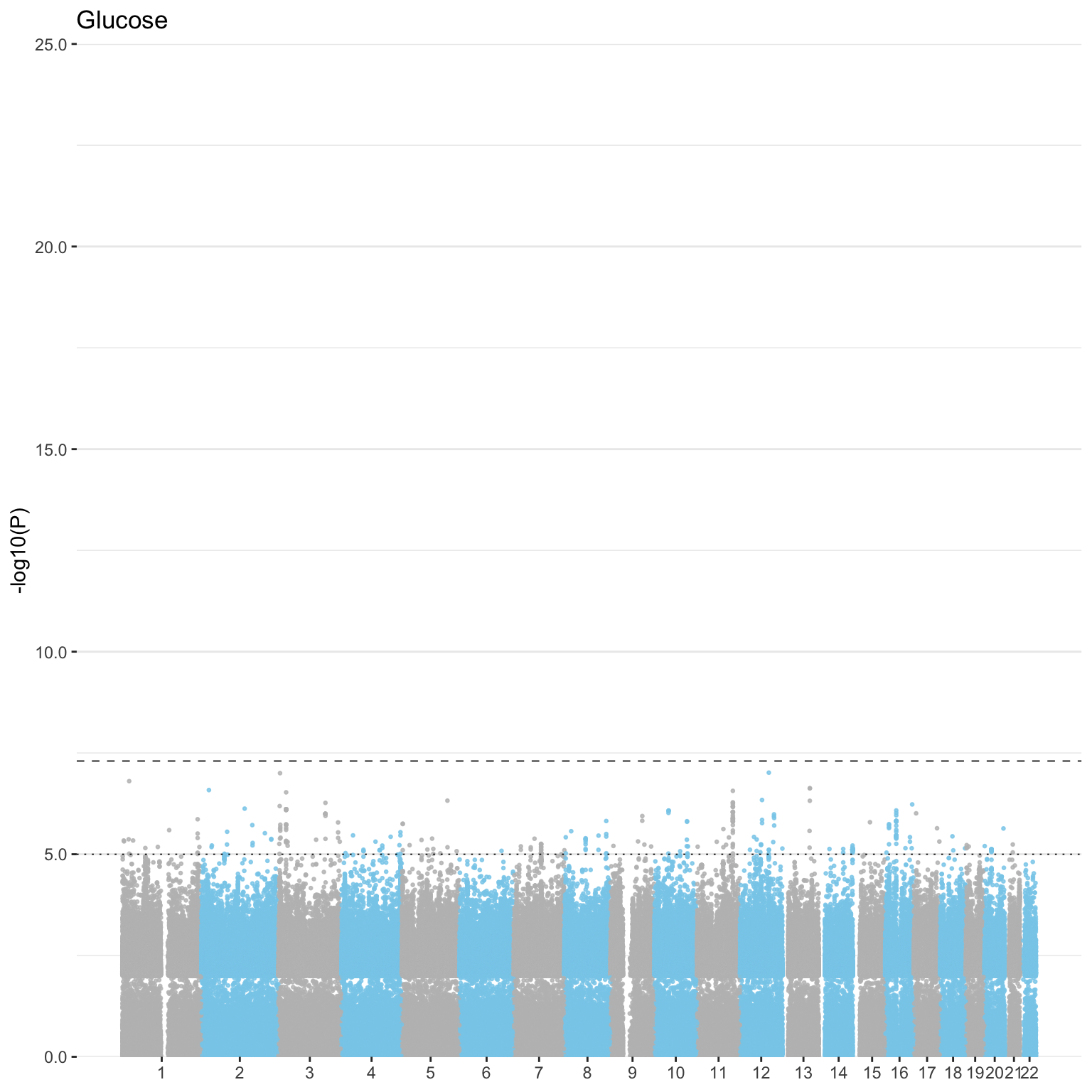
Ahora te toca a ti, realiza una gráfica similar para otro trait, en esta ocasión que el eje y no esté invertido y se muestre el número de cromosoma en el eje x.